Beyond the Total Plate Count: Metagenomic Applications for Meat and Poultry Processors
Advances in next-generation sequencing have allowed for an understanding of the microbial inhabitants that make up the microbiome in ways that culture-based techniques are limited

Today's commercial meat or poultry processing plant is a complex network of equipment, automation, human interaction, and multiple processes/pathways. These components work together to efficiently fabricate a carcass into multiple primal, subprimal, and trim cuts to be used for further processing. Here, they can be either packaged raw or further processed into ready-to-eat (RTE) meat items. At its core, this workflow has been successful for thousands of years in being able to create a wide variety of individual cuts of meat and further processed meat items such as sausage, which was noted as far back as Homer's Odyssey, written approximately 10,000 years ago.
While the processes have changed and advanced technology has been incorporated, one constant throughout the evolution of the meat industry has been the contribution of a very biologically diverse microbiome. This microbiome may contain thousands of different species of bacteria, viruses, and fungi living together as a community and taking residence in the animal, processing environment, and meat surfaces. The presence of specific bacteria within the microbial community can have both positive and negative effects on the safety and wholesomeness of the meat produced. The shelf life of both fresh and processed meat items is heavily influenced by the proliferation of select bacterial species within the meat microbiome under environmental conditions, which can result in undesirable characteristics of the product and a less-than-favorable experience for the consumer. Additionally, a critical focus of the meat and poultry industry has been the elimination of specific foodborne pathogens within the microbiome of the animal, plant environment, and ultimately the meat that is purchased by the consumer.
Meat and poultry processors have several tools to understand the context of the microbial ecology throughout the entire farm-to-fork supply chain. Culture-based techniques have been used by industry for decades and remain an important tool to get a good understanding of microbial load and the presence of selected pathogens. However, advances in next-generation sequencing have allowed for an understanding of the microbial inhabitants that make up the microbiome in ways that culture-based techniques are limited.
When given a toolbox full of tools, processors need to be able to determine which tool applies best to the research question that is being asked. Additionally, meat and poultry processors do not have unlimited research budgets to be able to answer these questions. A thoughtful, strategic approach to the overall goal of the testing can allow processors to select the most appropriate tool for the job. The objective of this article is to provide a broad overview of two genomic techniques and potential applications for their use by meat and poultry processors.
Where to Start?
Albert Einstein famously said, "If I had an hour to solve a problem and my life depended on the solution, I would spend the first 55 minutes determining the proper question to ask… for once I know the proper question, I could solve the problem in less than five minutes."
This approach certainly applies to meat and poultry processors as they first begin to establish what needs to be learned through testing. This is also the part of the process that tends to receive the least amount of attention when processors are faced with a crisis and answers are needed quickly. Having the discipline to clearly define the research question allows processors to determine the overall objective of the testing and will help avoid the trap of "testing for the sake of testing," which is often costly, labor-intensive for both the plant and the lab, and sometimes does not adequately answer the question. Once the research question is established, then an efficient test design and appropriate methods can be applied to answer the question.
Options to Answer, "Who is There?"
Several techniques present themselves as viable options when determining the number of bacteria and a broad understanding of which of them are present in the sample being taken. Culture-based techniques can provide insight into the consistency of the microbial population and, importantly, what can grow in the conditions being used in the lab. Strategically, starting broadly with culture-based testing will allow processors to understand the general trends within the microbial ecology of the environment or meat. Much can be learned from an aerobic plate count that can be helpful in making decisions around bacterial load changes. Additionally, observing colony morphologies and growth characteristics can give a general idea as to whether bacterial populations may be shifting, either over time or throughout a certain workflow. Culture-based methods are also economically viable and can be appropriate for routine monitoring activities.
Looking for quick answers on food safety topics?
Try Ask FSM, our new smart AI search tool.
Ask FSM →
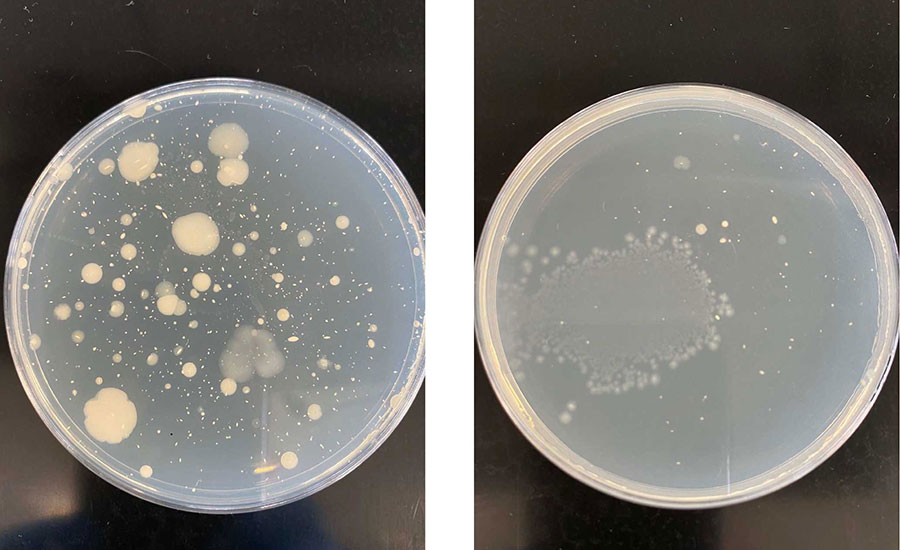
Figure 1 shows mesophilic aerobic plate counts of two different meat types sampled on the same day. The overall colony count from each plate can provide an idea of microbial load. The presence of different colony morphologies or growth characteristics can provide a broad idea of the overall microbiome diversity present in that sample.
However, metagenomic techniques can provide a deeper understanding of the overall microbial population present in the sample. Metagenomics refers to a culture-independent genetic analysis of the genomes present in a specified environment or sample. Since each organism present in the sample will have a specific genome, this approach can provide insight into the composition and diversity of the entire population and can also provide taxonomic identity to "Who is there?" within that sample, and how abundant each member is compared to the others. For the purposes of this discussion, there are two whole-sample, non-enriched, culture-independent methods that accomplish this: shotgun metagenomic sequencing and 16S sequencing.
Before proceeding, we will offer a quick refresher regarding taxonomy. All living organisms are classified based on shared physical, metabolic, and/or genomic characteristics. This classification is part of a hierarchal tiered system that begins at the broadest rank, domain, and progresses down in rank to kingdom, phylum, class, order, family, genus, and species. For example, Escherichia coli would be of the genus Escherichia and the species E. coli. Some bacteria can be further classified into subspecies and/or serotype. The pathogen Salmonella Enteritidis would be classified as Salmonella (genus) enterica (species) subspecies enterica serotype Enteritidis. The need to identify the lower taxonomical ranks is a key factor in determining whether to use shotgun metagenomic sequencing (more complex) or 16S sequencing (less complex and cheaper).
Shotgun metagenomic sequencing essentially works as the name implies. It involves the extraction of all DNA in a meat or environmental sample, the fragmentation of the DNA into smaller pieces (for short-read sequencing), and DNA sequencing that can occur using either a short-read or long-read platform. Sequencing provides a FASTQ file that contains so-called "reads," which represent the genetic code of each fragment of DNA from the original sample. This FASTQ file serves as the basis for the bioinformatic pipeline, which consists of a series of computer-based informatic tools and steps that, together, process the individual reads (i.e., sequences) in the FASTQ file. This pipeline may require a high degree of computing power and data storage to efficiently analyze the FASTQ files. The bioinformatic pipeline is often customized, but typically involves the removal of low-quality and host sequences (for example, Bos taurus DNA in the case of a ground beef sample), followed by assembly, alignment, and/or annotation of the sequences using existing genomic databases. These bioinformatic processes allow for the identification and characterization of sequences of interest, such as sequences from bacteria that might be involved in spoilage or virulence genes within known pathogens. Shotgun sequencing can provide taxonomic identification to the species or even strain level (if the data are large enough) and can also provide a broader view of the genes present in the sample. This can be valuable in understanding the relative abundance of genes associated with a trait such as antimicrobial resistance, or virulence factors within the sample or environment.
The method known as 16S sequencing is similar to shotgun sequencing in that it involves extraction of the total DNA from the sample. However, 16S sequencing involves PCR amplification of only the 16S rRNA gene and the sequencing of specific hypervariable regions within the 16S gene. The 16S rRNA gene is ubiquitous in prokaryotes (bacteria and archaea), but the hypervariable regions present within the 16S gene tend to have different sequences at the family or genus levels, and sometimes at the species level. In this way, the hypervariable regions of the 16S gene are like sequence "fingerprints" that can provide the taxonomic composition of the bacteria present in the sample. Since the 16S approach requires sequencing of only the very small, targeted, hypervariable regions of the 16S gene, it is generally less complex and cheaper to conduct than shotgun metagenomics sequencing, which requires sequencing of all of the DNA.
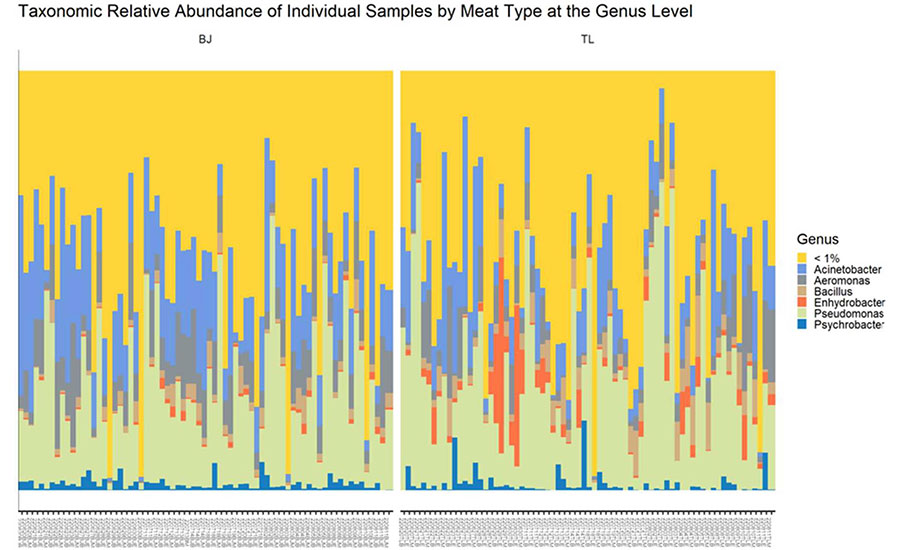
Figure 2 shows 16S taxonomic relative abundance separated by meat type. Each bar represents an individual meat sample, with different colors within the bar designating a unique genus identified within the microbiome data. Developing a chart like this can be helpful when comparing the presence of certain genera within a sample between two meat types, the meat vs. contact surface, or comparing different points in the fabrication process such as bio-mapping of lairage through the carcass cooler. This chart reflects the 16S relative abundance of two different meat types (BJ and TL). Each vertical bar represents an individual sample taken from the BJ and TL. This chart shows that there is high sample-to-sample variability in the makeup of the microbiome within the two meat types and fabrication processes, even from samples taken within the same processing plant. It also shows that a large proportion of the meat microbiome is made up of low-abundance taxa, which are shown in yellow; these low-abundance taxa each represent < 1 percent of all of the genomic data in the sample.
For meat and poultry processors, 16S sequencing can provide valuable insight toward understanding the microbial ecology of the meat or environment in a relatively efficient and cost-effective way. While a culture-based aerobic plate count can give an idea of load and morphologic diversity, 16S sequencing can provide an answer to "Who is there?" in the plate count, including the bacteria that may not be culturable or distinguishable with plating methods. It is important to note that 16S sequencing is limited in that it will not provide species- or sometimes even genus-level identification for all bacteria identified in the sample, and it cannot distinguish between closely related genera. It also does not inform which genes or functions are present within the bacterial genomes. While species and gene information can be provided by shotgun metagenomic sequencing, this comes at an increased cost and complexity. A specific question and requirement that would warrant shotgun metagenomics would need to be evaluated in the initial project design.
Applications of 16S and Shotgun Metagenomic Sequencing
Several applications exist where metagenomic sequencing can be a valuable tool for meat and poultry processors, especially when used in complement with traditional, culture-dependent methods. An understanding of the plant environment microbial ecology can provide processors with information to optimize sanitation efforts, determine the effect of interventions beyond the prevalence of pathogens, and provide insight into how these changes may affect the shelf life of the finished meat product. Again, the decision to use either 16S or shotgun metagenomic sequencing depends on the research question being asked and whether or not it is necessary to attain the identification of bacteria to the species level and/or to understand the phenotypic potential of the genes present in the sample.
Shotgun sequencing and 16S can help provide insight to plant environment research questions such as:
- How does the meat or carcass microbiome change with exposure to harvest and fabrication processes in a production environment?
- Is the presence of certain antimicrobial resistance genes in the meat associated with the microbiome of the live animal, or are these genes additively acquired from other bacteria throughout the harvest/fabrication process?
- Is there an association between the presence of specific bacteria on the carcass and specific bacteria present in individual meat cuts or trim?
- Is the microbiome of each primal cut unique by fabrication line?
- Does the microbiome diversity of the meat or environmental contact surface change throughout the production day, week, or with changes in season or weather?
- Does fabrication equipment, process, or carcass anatomy contribute to the diversity of the meat or contact surface microbiome?
- Is there an association between the presence of certain pathogens and the presence of specific spoilage bacteria in the meat or plant environment?
Additionally, metagenomic sequencing can be a valuable tool in understanding how the microbiome of the finished meat product changes throughout the duration of shelf life and in understanding how contributions of the plant environment, interventions, or formulated antimicrobials may affect shelf life. While many processors have an established maximum count of either mesophilic or psychrotrophic bacteria that determines the end of shelf life, it is not always a fail-proof indicator of the growth of specific bacteria that contribute to physical failure characteristics such as off odors, flavors, or unacceptable package appearance. Using either 16S or shotgun sequencing as a complement to culture-dependent analyses can provide insight into how the overall relative abundance of certain bacteria within the sample are changing over the course of shelf life. This can provide processors with the ability to address the specific bacteria driving the shelf life of the product, potentially through sanitation or interventions at the processing plant or through the formulation of an antimicrobial system that effectively targets the organism of concern.
It is important to note that the results of 16S or shotgun sequencing analysis are compositional in nature and will provide a relative abundance of the bacteria present. For example, the total number of reads (sequences) that originate from a certain bacterial genus would be summed to estimate the relative abundance of that genus in the sample. This relative abundance number can be associated with the actual count of that specific genus, but this count is not an absolute value and, therefore, does not provide a direct measure of the load of that genus in the sample. This is also why nonselective plate count methods can be an excellent complement to a metagenomic research project, by providing enumeration and giving better context to the relative abundances calculated from the metagenomic analyses.
Additionally, statistical measures for alpha diversity (the diversity within a sample) and beta diversity (similarity/dissimilarity compared between samples) are commonly used to analyze and visualize the diversity and composition of microbial populations within and across samples; again, culture-based approaches can augment these analyses and provide more context about the microbial ecology. To fully leverage these types of datasets, processors will need to familiarize themselves with these statistical measures as they are developing the research question and test.
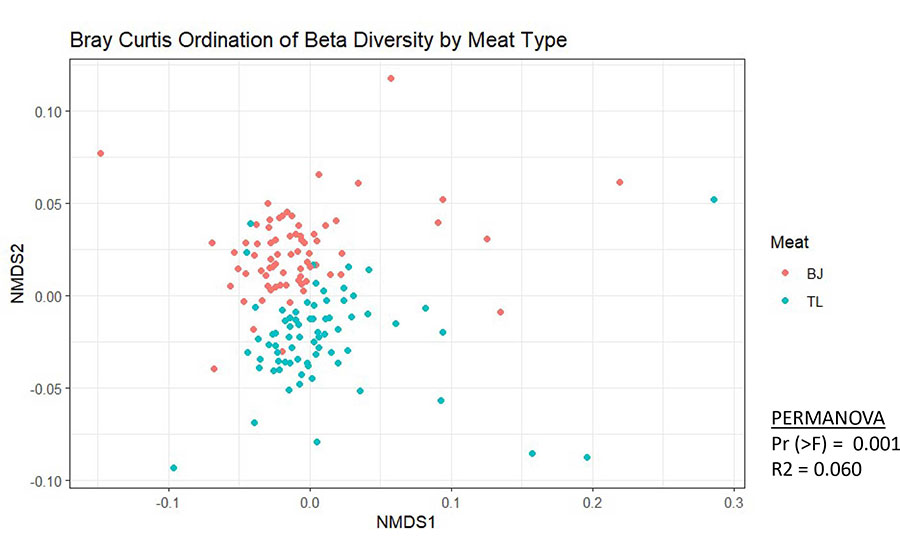
Each dot in Figure 3 represents the beta diversity of a single sample, comparing two different meat types (represented by red and blue dots) based on the 16S data from the sample. Dots that are closer together have a more similar microbiome composition than do dots that are further apart. In this chart, clustering between samples/dots of the same meat type suggests a more similar overall microbiome within each of the meat types. Visualizing beta diversity like this can be used to compare the similarity of the microbiome between two meat types, between the carcass surface pre- and post-application of a surface intervention, between the meat and contact surfaces, between two plants that make the same product (with one failing shelf life), or between a product formulated with and without a certain antimicrobial. These plots are a convenient way to understand shifts or differences in the microbial populations of different types of samples or the same types of samples subjected to different interventions or environmental conditions.
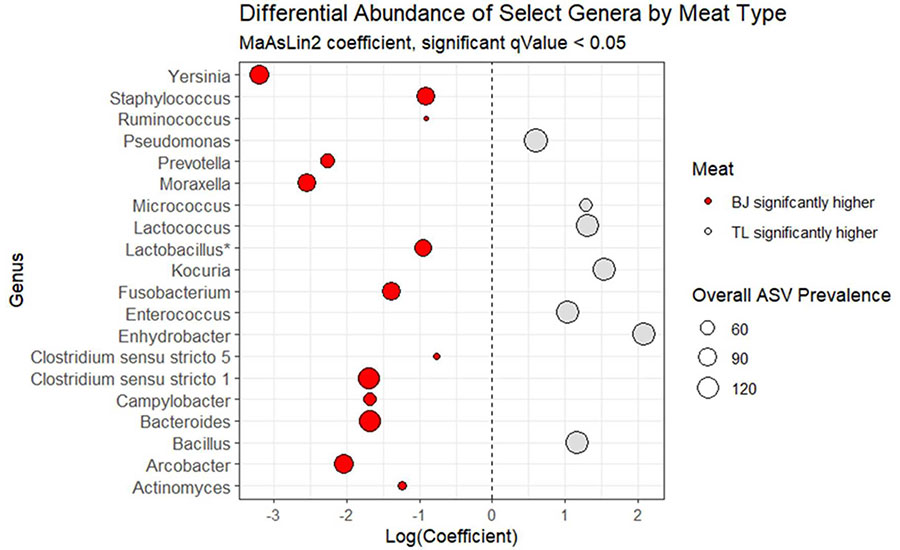
In Figure 4, the different-colored dots reflect genera that are significantly more abundant between the meat types analyzed. The size of the dot reflects the overall prevalence of each key genus observed within the study, and the Log(Coefficient) (x-axis) reflects the magnitude of the difference, in this case between two different meat types. Differential abundance can be useful in understanding which bacterial genera may be driving the overall shelf life of a meat product; whether the presence of key bacteria may change throughout a production shift; or if the presence of key bacteria may be associated with a particular meat type, fabrication process, or presence of a pathogen.
Where is This Going?
The evolution of next-generation sequencing has substantially reduced the cost of conducting a genomics-based research study. Relatively affordable entry points for sequencing platforms are available. However, owning a sequencer is just a starting point to be able to conduct a genomic study. While the analysis is DNA-based, the successful ability to process the sample, extract the DNA, and provide the sample to the sequencer is still driven by lab technicians, who must be proficient in molecular techniques, including best-practice techniques that minimize contamination and bias that may affect the results. Additionally, genomic analysis requires extensive computational capabilities, data storage, and may also require hiring additional staff that have a background in bioinformatics and microbiome analyses, specifically. For these reasons, it may be difficult for most meat processors to build these capabilities internally and be able to justify the initial startup costs unless a definitive, ongoing research need is determined.
However, opportunities exist to partner with either third-party laboratories or university researchers to conduct both exploratory and hypothesis-based metagenomic research in the plant. These laboratories already have the expertise and hardware necessary to conduct the extraction, sequencing, bioinformatics, and data analysis. As is the case with any research project, the overall research question, test design, and roles of the meat processor and external lab/university must be clearly defined and understood before initiating the project.
As the cost of sequencing continues to decrease, more applications for its use will be determined and the financial and computational barriers will be overcome. Culture-independent methods certainly have a place in the toolbox for processors to understand the plant environment, meat, and finished product shelf life in situations where culture-dependent analyses may be limited. Further research and development of target-enriched, long-read sequencing may also provide a pathway for culture-independent diagnostics that can identify the presence and sequence of low-level pathogens in a meat or environmental sample. The potential exists for these techniques to be used on a more regular basis, especially as food safety discussions start to move from overall prevalence to load, and then potentially to the genes present.
As with any tool, the utility of the tool is often determined by the job it is being asked to do. Scalpels can be useful when the job requires fine detail, but no one is going to use a scalpel to chop down a tree. As such, shotgun metagenomics and 16S sequencing can be very useful tools when the need arises. Meat and poultry processors should become familiar with the capabilities of each and assess potential applications for their use to better understand the microbial ecology of their processing plants, or as a complement to their current environmental monitoring or shelf-life programs.
Aaron Asmus, M.Sc., is the Director of Lab Services and Refrigerated Foods Product Development at Hormel Foods Corporation in Austin, Minnesota. He is also a Ph.D. candidate at the University of Minnesota College of Veterinary Medicine in St. Paul, Minnesota.
Noelle Noyes, Ph.D., D.V.M., is an Assistant Professor at the College of Veterinary Medicine at the University of Minnesota in St. Paul, Minnesota.








